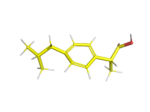
This is the second post in a series aiming at generating a range of candidate structures for evaluation in the […]
This is the first of a series examining the use of python to generate candidate structures of molecules. These conformations […]