This is the 3rd post in a series outlining a workflow using freely available computational chemistry resources with python interfaces […]
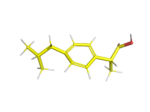
This is the second post in a series aiming at generating a range of candidate structures for evaluation in the […]